В эпоху больших данных получить огромное количество информации несложно; знать, что с этим делать, это совсем другая история. Но теперь исследователи из Японии сообщили, что новый подход к анализу данных полногеномных ассоциативных исследований может помочь раскрыть генетическую основу многих заболеваний.
В исследовании, опубликованном в августе в Nature Communications , исследователи из Токийского медицинского и стоматологического университета (TMDU) показали, что анализ кодирующих последовательностей вариантов сплайсинга генов в участках, связанных с заболеванием, может помочь выявить генетическую причину некоторых сложных заболеваний человека.
Вариации в наших генах вызывают сложные заболевания, но может быть трудно сказать, как одна генетическая вариация приводит к заболеванию. В то время как некоторые варианты вызывают заболевание, изменяя уровни экспрессии генов , становится все более очевидным, что варианты сплайсинга, которые влияют на то, как транскрибируется ген, то есть на то, как последовательность ДНК гена копируется в РНК, также играют важную роль.
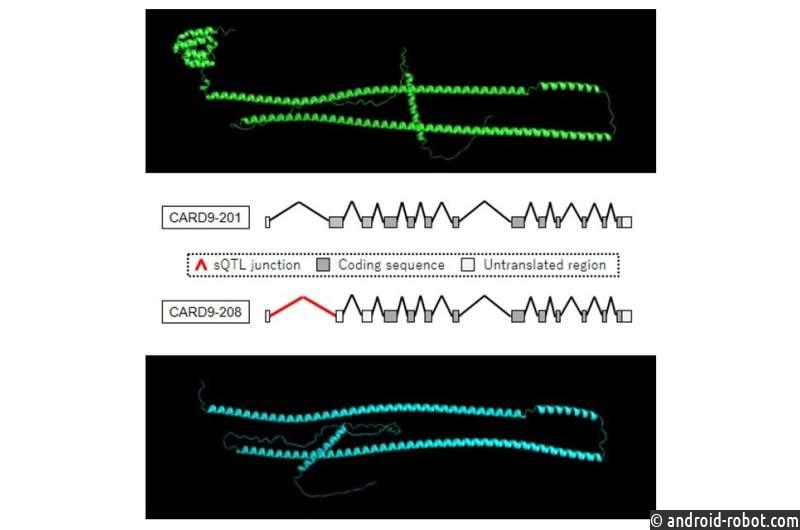
«Существует ряд существующих подходов для выявления и анализа генетических вариантов, вызывающих сплайсинговые изменения в генах, связанных с заболеванием», — объясняет Кенсуке Ямагучи, ведущий автор исследования. «Однако эти подходы ограничены неполной аннотацией изоформ сплайсинга и использованием одного и того же соединения сплайсинга несколькими изоформами, из-за чего их трудно отличить друг от друга».
Чтобы преодолеть эти недостатки, исследователи разработали набор из двух анализов, которые более полно отражают сложность вариаций сплайсинга и их связь с болезнями человека : первый анализ объединяет изоформы с одной и той же кодирующей последовательностью для обнаружения результирующих изменений в структуре белка , а второй анализ исследует влияние изоформ с неполными аннотациями, но уникальными кодирующими последовательностями. Затем команда определила полные последовательности этих изоформ и проверила их экспрессию в клетках.
«Результаты показали, что наш подход является надежным и эффективным», — говорит Юта Кочи, старший автор статьи. «Мы успешно идентифицировали 29 полноразмерных изоформ с неаннотированными кодирующими последовательностями, связанными с генетическими вариантами, которые были связаны с такими заболеваниями, как болезнь Паркинсона, анкилозирующий спондилит, заболевание раздраженного кишечника и нейродегенеративное заболевание».
Кроме того, они показали, что гены с вариантами сплайсинга, ассоциированными с заболеванием, могут быть идентифицированы путем оценки их влияния на экспрессию других генов в геноме. Например, вариант , приводящий к изменению соотношения двух изоформ гена SNRPC, был идентифицирован как ассоциированный с системной красной волчанкой .
Взятые вместе, эти данные подчеркивают недооцененную роль вариантов сплайсинга, изменяющих белок, в возникновении заболевания. Выявление соответствующих вариантов и оценка их функции в будущих исследованиях с использованием моделей на животных может помочь прояснить, как возникают сложные заболевания.