Метилирование ДНК — это биологический процесс, посредством которого к ДНК (генетическому материалу) добавляются метильные группы. Он используется прокариотами в качестве эпигенетической, т. е. негенетической стратегии для выполнения множества функций, таких как регуляция генов, репарация и защита от вирусной инвазии с использованием систем рестрикции-модификации (RM), которые функционируют как прокариотические иммунные системы.
До недавнего времени исследования, связанные с метилированием ДНК, ограничивались микроорганизмами, которые можно культивировать в лабораторных условиях. Это привело к плохому пониманию его роли в микробной экологии. Поэтому крайне важно проводить полногеномные эпигенетические исследования микробов окружающей среды, особенно тех, которые не могут быть культивированы в лаборатории, а процветают только в естественных условиях.
С этой целью группа исследователей под руководством профессора Ву Джун Сула из Университета Чунг-Анг и доктора Хун Дже Сонга (в настоящее время из Macrogen Inc.), Южная Корея, изучила различия в моделях метилирования ДНК у разных представителей океанических микробов. сообщества в северо-западной части Тихого океана.
Их исследование было опубликовано в Microbiome.
«[Проект] углубленного метилирования ДНК начался только в 2014 году, когда были выпущены долгочитаемые секвенаторы. Это вызвало у нас любопытство, и мы захотели применить его к микробной экологии. Поэтому мы использовали метагеномный подход для изучения метилирования ДНК в сообщества, а не на уровне организма», — говорит профессор Сул, обсуждая мотивы своего исследования.
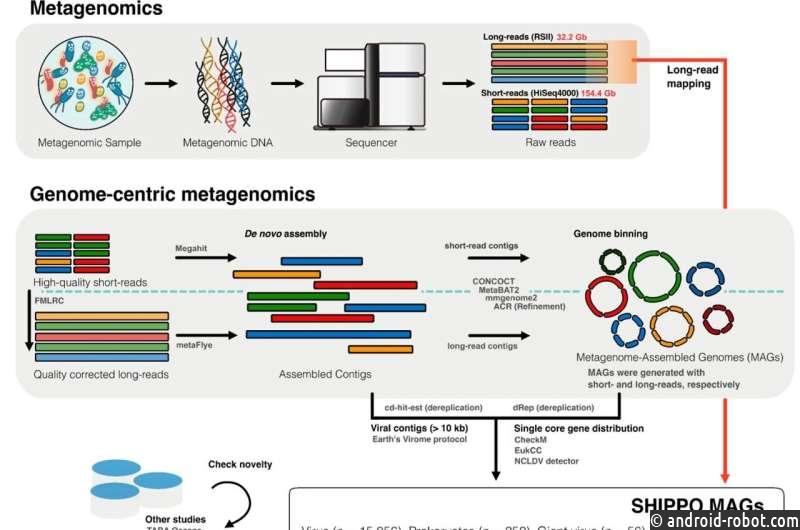
Ажиотаж начался еще в 2015 году, когда Корейский институт полярных исследований инициировал крупномасштабный проект морских наблюдений от полюса к полюсу (SHIPPO). Он включал фильтрацию микроорганизмов из образцов поверхности океана на 10 различных станциях от тихоокеанского северо-запада до Берингова моря.
Команда извлекла ДНК из этих захваченных образцов и использовала секвенаторы с коротким и длинным считыванием для выполнения метагеномного секвенирования. Затем эти последовательности были выровнены с помощью компьютерного анализа для создания массивных 15 056 вирусных (v), 252 прокариотических (pro), 56 гигантских вирусных (gv) и 6 эукариотических (eu) геномов, собранных метагеномом (MAG).
При дальнейшем анализе команда с удивлением обнаружила, что почти 95% секвенированных proMAG принадлежали к новым таксонам, которые не могли быть классифицированы с использованием существующих геномных баз данных. «Это открытие ясно демонстрирует потенциал этого метода и то, как он может дать новое представление о геномах некультивируемых океанских микробов», — объясняет профессор Сул.
Затем команда использовала этот подход для изучения разнообразия классов ферментов ДНК-метилтрансферазы (МТазы), экспрессируемых геномами, идентифицированными в базе данных SHIPPO.
Они обнаружили, что МТаза II была наиболее распространенным классом МТазы, экспрессируемой в этих организмах. Интересно, что в большинстве proMAG отсутствуют полные системы RM из-за отсутствия ферментов рестрикции. Кроме того, идентификация метилированных мотивов в микробиоме океана выявила уникальные модели метилирования ДНК, что в конечном итоге привело к открытию особого профиля метилирования у альфапротеобактерий.
Затем команда использовала секвенирование отдельных молекул в реальном времени (SMRT) для наблюдения за паттернами метилирования у Pelagibacter. Они обнаружили гетерогенность в профиле метилирования бактерий даже на «штаммовом уровне». Это означает, что динамические клеточные события происходят внутри Pelagibacter в поверхностных водах северо-западной части Тихого океана.
Сравнительный анализ геномов бактерий и вирусов также дал ключ к разгадке их эволюционных моделей и взаимодействий. Команда обнаружила наличие неравномерных паттернов метилирования в Cand. Геном P. Giovannoni NP1, что предполагает потенциальные защитные механизмы, используемые этой бактерией.
Эти открытия уже проложили путь к новой эре метаэпигеномики, которая напрямую измеряет метилирование микробов окружающей среды. Потенциал изучения эпигенома различных организмов одновременно является далеко идущим.
Профессор Сул говорит: «Наряду с исследованиями по выявлению паттернов метилирования штаммов, проявляющих реальную патогенность, наше исследование также помогает обнаружить мишени-кандидаты для предотвращения патогенности в окружающей среде. Это может иметь огромное значение для глобальных систем общественного здравоохранения, обнаруживая патогенные сигналы, которые угрожают здоровью человека».