В последние годы исследования и разработки в области квантовых компьютеров значительно продвинулись вперед. Квантово-химические расчеты электронной структуры атомов и молекул привлекают большое внимание как одно из наиболее многообещающих приложений для квантовых компьютеров. Чтобы использовать квантово-химические расчеты для химии и смежных областей, необходимо разработать методы оптимизации геометрии, чтобы найти наиболее стабильную структуру для молекул. Оптимизация геометрии требует вычисления производных энергии по ядерным координатам молекул.
Метод конечных разностей является одним из подходов к расчету производной энергии. На классическом компьютере расчеты по этому методу для одномерных систем требуют не менее двух оценок энергии. Предыдущие исследования показали, что квантовому компьютеру, напротив, требуется только один запрос для вычисления производных энергии на основе метода конечных разностей, независимо от количества степеней свободы. Однако квантовые схемы , относящиеся к квантовым алгоритмам, способным выполнять вычисления производных энергии, не реализованы.
Исследовательская группа, включающая доктора Кендзи Сугисаки, профессора Казунобу Сато и почетного профессора Такеджи Такуи из Высшей школы наук Университета Осаки Метрополитан, успешно расширила алгоритм оценки квантовой разности фаз, общий квантовый алгоритм для прямых расчетов энергетических щелей. чтобы обеспечить прямой расчет разницы энергий между двумя молекулами с разной геометрией. Это позволяет вычислять на основе метода конечных разностей производные энергии по ядерным координатам за один расчет.
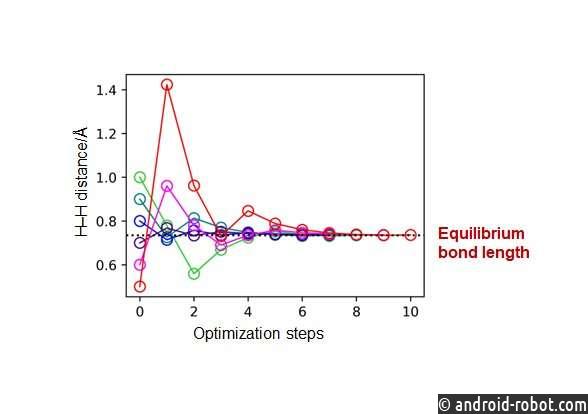
Кроме того, исследовательская группа применила разработанные расчеты производных энергии для оптимизации геометрии молекул H 2 , LiH, BeH 2 и N 2 без расчета полных энергий, демонстрируя полезность разработанного метода. Группа также обсудила, как квантовые схемы могут быть собраны в соответствии с различными степенями свободы молекул.
Это исследование является последним в серии статей исследователей о квантово-химических расчетах на квантовых компьютерах. «Наши последние результаты приближают нас на один шаг к применению квантово-химических расчетов на квантовом компьютере для решения реальных проблем», — сказал доктор Сугисаки.
«Поскольку расчеты производной энергии используются не только для оптимизации молекулярной геометрии, но и для различных расчетов молекулярных свойств, ожидается, что применение нашего метода сыграет очень важную роль в широком спектре смежных областей, таких как открытие /разработка силиконовых лекарств и разработка материалов».